
Current Highlights
Review of CRASY Achievements: In 10 years of CRASY spectroscopy, we measured mass-correlated spectra for numerous isotopologues at natural abundance, determined gas-phase molecular structures with mA resolution, characterized unexpected cationic fragmentation channels, characterized molecular cluster spectra, and developed the highest-resolution scanned interferometric measurement method in the world. Read all about these topics in a review that will soon appear in PCCP. [arXiv:2403.03634]
Correcting the Benzene Structure: Spectroscopic results for benzene isotopologues allowed us to determine precise effective and equilibrium geometries. Unfortunately, our results contradicted literature values for equilibrium geometry bond lengths and geometric isotope effects. Read our work to learn about the correct benzene structure and why everybody else got it wrong. [RSC Adv., 2022, 12, 21406][JPCL 2022, 13, 8278]
Highest resolution rotational Raman spectra: Recent mass-selected rotational spectra obtained with CRASY reach a resolution of 0.3 MHz and are the highest resolution Raman spectra in the literature measured over broad spectroscopic bandwidth. The resolution is based on the observation of rotational wave packets over a time range beyond 1 microsecond. [Phys. Chem. Chem. Phys., (2019)] We explained the technology behind these measurements in detail in our recent paper “Molecular-beam spectroscopy with an infinite interferometer: spectroscopic resolution and accuracy”. [J. Korean Phys. Soc. 82(9), 919 – 927 (2023)]
Highly-accurate rotational constants: Rotational frequencies measured by CRASY can be calibrated against an external frequency standard with a simple measurement of the oscillator repetition rate. The resulting measurement represents a first time-domain equivalent to frequency comb spectroscopy (cf. 2005 Physics Nobel prize for R. Glauber, J. Hall, and T. Häntsch). [Proc. Natl. Acad. Sci. U. S. A., 115, pp 5072 (2018)]. Rotational constants obtained by CRASY for the molecules carbon disulfide, benzene, butadiene, and their isotopologues all reached an accuracy of Dn/n < 10-6 and established accurate rotational constants for each molecule.
Dual amplifiers and time delay range from few nanoseconds to microseconds: A single femtosecond oscillator seeds two separate regenerative amplifiers. Each amplifiers (for pump and probe) can independently amplify selected pulses in the pulse train. Time delay between pump and probe is adjusted with opto-mechanical (OM) and electronic pulse-selection (PS) for continuous scanning from femtoseconds to microseconds.
Sparse sampling: A better frequency resolution requires a longer sampling time. To simultaneously obtain a large frequency range (=small sampling step size) and a high resolution (=long sampled delay range) would result in a tremendous experimental cost in terms of time. Hence, random sparse sampling is performed to expedite long measurements.
Broad spectral bandwidth: To maintain a large spectroscopic range, we randomly sampled delay values in multiples of 1 ps (The maximal frequency is μmax = 1/{2(delay_stepsize)}), leading to a Fourier-domain spectral range of 0 to 500 GHz. μmax =

Going beyond the limits of traditional (ultrashort pulse) spectroscopy
Electrons and ions formed in molecular ionization can be detected with quantum yields close to one, leading to an extraordinarily sensitive characterization of molecular mass and electronic structure. By using pump-probe ionization schemes, the characterization can be extended to photo-excited species to observe photochemical reactions in molecules and clusters [1-4]. The information content of such experiments, however, is insufficient for a spectroscopic assignment of molecular structure in all but the most trivial molecules.
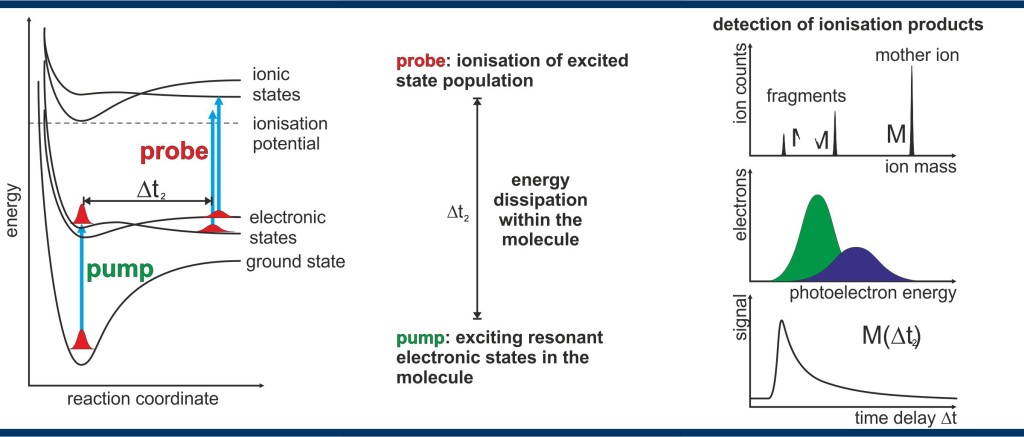 Figure – Femtochemistry:
Figure – Femtochemistry:

Left: energy scheme to illustrate a standard pump probe experiment to measure energy dissipation in a molecule
Center: a pump step excites electronic states – within a time delay, energy dissipation in the molecule occurs – the electronic state population is probed by ionization
Right: ionization products can be detected with quantum yields close to one – a mass spectrum identifies mother ions and their fragmentation products – an electron spectrum identifies electronic states involved in the resonance enhanced ionization of the molecule – signals as function of time delay Δt2 reveal photochemical reaction pathways.
We presented the spectroscopic method of correlated rotational alignment spectroscopy (CRASY) to overcome this limitation [5]. The additional element, compared to traditional femtosecond pump-probe experiments, is a third IR-laser pulse which pre-excites a rotational wave packet in the molecular ground state before traditional experiments are performed. This allows CRASY to correlate (simultaneous) measurements of rotational structure, molecular mass, and electron binding energy for multiple molecules in a sample.
 Figure – Rotational spectroscopy in the time domain (characterization of the rotational wave packet):
Figure – Rotational spectroscopy in the time domain (characterization of the rotational wave packet):

The characterization of the rotational wave packet can be described as pump-probe experiment as well. A pump pulse (ps-IR) is exciting a coherent superposition of rotational states by means of Raman excitation. A wave packet is created. The state population remains in the molecular ground state.
The evolution of the wave packet can be visualized through a probe pulse (<< ps) by making use of electric dipole transitions. Transition dipole moments are fixed in the nuclear frame of a molecule. Hence, rotating molecules result in rotating transition dipole moments. The orientation of every transition dipole moment in a probing laser field varies as a function of time delay Δt1. The excitation probability correspondingly changes. The excited state population is modulated by the rotational motion of the molecular ensemble. Rotational frequencies are encrypted in the pump-probe signal.
Ionization projects the excited state population onto ionic states. For this reason, the time dependent ion- and electron signals reflect the evolution of the rotational wave packet in the ground state. A Fourier transformation discloses the rotational Raman spectrum of the excited molecule.
Info: The coherence time of the rotational wave packet is determined by radiative relaxation, which is in the order of seconds. Coherence will be lost if collisions occur. Vacuum spectrometers are therefore well suited to study rotational coherences in molecular ensembles.
The method of CRASY is based on the coherent Raman excitation of a rotational wave packet with a strong infrared laser-pulse (Figure – Rotational spectroscopy in the time domain). The excitation probability in a subsequent pump-probe ionization experiment is modulated by the rotation of the molecules and leads to Δt1 time-dependent signal modulations in electron and ion signals. A Fourier transform of the temporal signal modulations gives frequency-domain rotational Raman spectra for all detected masses and electron-energies. Therefore, the ground state rotational Raman spectrum is correlated with all observables of pump-probe ionization spectroscopy. This way CRASY combines the high sensitivity and selectivity of mass spectroscopy with the large information density of high-resolution rotational spectra. Additional variation of the time-delay Δt2 between pump and probe pulses allows to determine excited state lifetimes. Together with the observation of transient electronic structure via electron-spectroscopy, CRASY therefore opens the door for the structure-selective assignment of photochemical processes even in impure or instable samples.
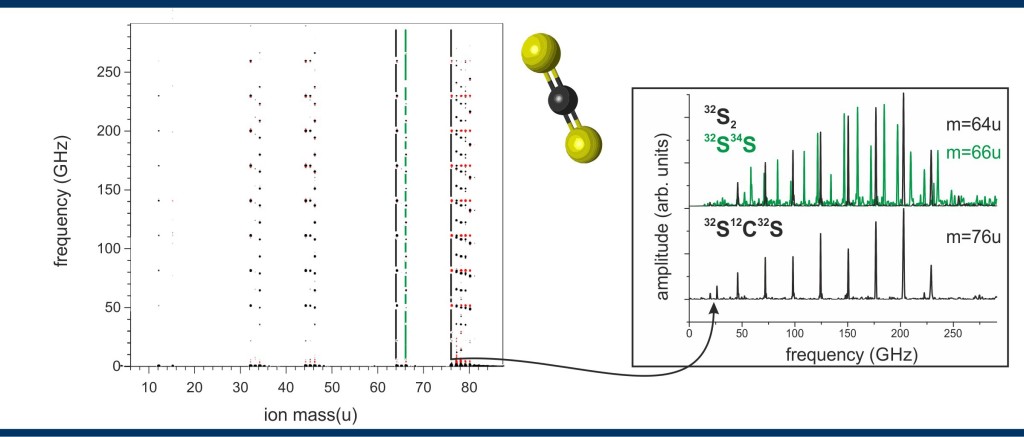 Figure – Two-dimensional data set of carbon disulfide (2011 data):
Figure – Two-dimensional data set of carbon disulfide (2011 data):

left: The two-dimensional data received with mass-CRASY is spanned by the frequency and ion mass axis. Every signal in the map corresponds to a rotational Raman transition frequency linked to a specific ion mass detected with a time-of-flight mass spectrometer. The information content, e.g., allows to identify fragmentation products of specific structural species inside the sample.
right: As an example the fragmentation products containing two sulfur isotopes are shown. The spectrum extracted for the 34S32S (m=66u) fragment clearly differs from the spectrum obtained for the 32S2 (m=64u) fragment. Hence, both fragments belong to different parent molecules as obvious in this example. The spectrum for mass channel 76u shows the rotational transition frequencies for the 32S12C32S isotopologue, the mother ion of the sulfur dimer fragment.
The large information content of a mass-CRASY data set is illustrated with data from a single experiment on CS2 (Figure – two-dimensional data set of carbon disulfide). Multiple mass signals in the range m/e 76-82 are due to the presence of 12C, 13C, 32S, 33S, 34S, and 36S isotopes. In some cases, the isotopic composition is reflected in distinct masses (e.g., 32S12C32S versus 32S13C32S), in other cases it changes the molecular moments of inertia and leads to distinct rotational spectra (e.g., 32S13C32S versus 33S12C32S). The resonant two photon ionization process leads to fragmentation into molecular and atomic fragments with observed ion masses of 64-68u (S2), 44-48u (CS), 32-34u (S) and 12-13u (C). A vertical cut through the data at a selected mass yields the rotational-Raman spectrum of the neutral molecule before ionization. Each spectrum contains information on the molecular moments of inertia and the relative angle of transition dipoles and thereby offers the required fingerprint for the assignment of molecular structure. The displayed data set allowed the assignment of rotational constants for 10 naturally occurring CS2 isotopes with abundances down to 0.000002. The ability to generate mass-selected rotational spectra with very high sensitivity will be useful for the structure determination of low-abundance compounds in impure samples.
The available rotational resolution in CRASY experiments is crucial to allow the investigation of larger biomolecules and of structurally complex clusters. The resolution is primarily limited by the length of the available delay-line for Δt1 scanning. With a folded 16ns delay line, we increased the available resolution to below 65 MHz (Figure – Increasing the frequency resolution). This will be sufficient to (i) measure rotational spectra for low-abundance compounds in impure and instable samples, (ii) characterize electronic structure and electronic dynamics for inseparable molecular isomers, (iii) investigate electronic structure and fragmentation pathways of (bio-) molecular clusters, and (iv) measure rotational spectra of isotopologues.
 Figure – Increasing the frequency resolution (2009~2019):
Figure – Increasing the frequency resolution (2009~2019):

Observing the rotational motion of molecules over a certain period allows for the determination of rotational frequencies. The longer the rotational motion can be observed, the more precise the rotational frequency can be determined.
Published data from 2011 [5] was recorded with an opto-mechanical (OM) delay stage of 30 cm in September 2009. The observation time was therefore close to 2 ns. This results in a frequency resolution of 500 MHz. In the end of the year 2011, a beam folding setup was added to the delay unit, allowing for delays up to 16 ns. This directly converts to a total frequency resolution of 62.5 MHz, gaining almost an order of magnitude in frequency resolution.
After moving laboratory equipment to Korea, our group set up a laser system which allows to go far beyond the already obtained frequency resolution of 62.5 MHz. A single femtosecond laser oscillator (80 MHz repetition rate) seeds two separate regenerative amplifiers (1 kHz repetition rate). Each of the amplifiers can independently amplify selected pulses from the oscillator. This way, an OM delay can scan from one oscillator pulse to the next one, before the delay can be increased by another 12.5 ns (1 / 80MHz) via an electronic pulse-selection (PS). Note that 12.5 ns is the distance of two oscillator pulses in the pulse train. A frequency counter monitors the oscillator period against a GPS-stabilized external clock and characterizes the the discrete delays with high accuracy. This allows for pulse delays up to μs with femtosecond accuracy. The achievable frequency resolution is therefore not limited by the optical setup, but by other experimental aspects.
The measurement is performed in a molecular-beam mass spectrometer. Vaporized molecules were released through a pulsed valve and the expanding molecular beam was skimmed and arrived in a Wiley-McLaren mass spectrometer, where the molecules interacted with the laser pulses.

Table – Resolution of high-resolution spectroscopy techniques
The CRASY rotational resolution now exceeds the resolution of any preceding Raman spectroscopic method by more than an order of magnitude.
With the demonstration of the first CRASY experiments, we stand at the beginning of a new era for gas-phase spectroscopy. Even without the ability for structural discrimination, mass spectrometry became one of the most relevant technologies for molecular characterization in biology and medicine. The correlation of multiple spectroscopic observables, and in particular the ability to distinguish (and eventually to assign) structural isomers, will increase the information content of such gas-phase experiments by orders of magnitude. In the future, a careful choice of correlated observables will allow the analysis of complex molecular systems that were inaccessible to traditional (uncorrelated) techniques.
[1] A. Stolow, A.E. Bragg, and D.M. Neumark, ‘Femtosecond Time-Resolved Photoelectron Spectroscopy’, Chemical Reviews 104, 1719 (2004). [2] I.V. Hertel and W. Radloff, ‘Ultrafast Dynamics in Isolated Molecules and Molecular Clusters’, Reports on Progress in Physics 69, 1897 (2006). [3] K. Kosma, C. Schröter, E. Samoylova, I.V. Hertel, and T. Schultz, ‘Excited-State Dynamics of Cytosine Tautomers’, J. Am. Chem. Soc., 131, 16939 (2009). [4] E. Samoylova, W. Radloff, H.H. Ritze, and T. Schultz, ‘Observation of Proton Transfer in 2-Aminopyridine Dimer by Electron and Mass Spectroscopy’, J. Phys. Chem. A, 113, 8195 (2009). [5] C. Schröter, K. Kosma, and T. Schultz, ‘CRASY: Correlated Rotational Alignment Spectroscopy’, Science 333, 1011 (2011). [6] Christian Schröter, Chang Min Choi and Thomas Schultz, ‘CRASY: Correlated Rotational Alignment Spectroscopy Reveals Atomic Scrambling in Ionic States of Butadiene’, J. Phys. Chem. A, 119, pp 1309 (2015). [7] Christian Schröter, Jong Chan Lee and Thomas Schultz, ‘Mass-correlated rotational Raman spectra with high resolution, broad bandwidth, and absolute frequency accuracy’, Proc. Natl. Acad. Sci. U. S. A., 115, pp 5072 (2018). [8] Jong Chan Lee, Dong Eun Lee and Thomas Schultz, ‘High-resolution rotational Raman spectroscopy of benzene’, Phys. Chem. Chem. Phys., , (2019). [9] Weber, A. In Handbook of High-resolution Spectroscopy; Quack, M., Merkt, F., Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2011; Vol. 2; Chapter High-resolution Raman Spectroscopy of Gases, pp. 1153ff. [10] Frey, H.M. In Handbook of High-resolution Spectroscopy; Quack, M., Merkt, F., Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2011; Vol. 2; Chapter High-resolution Rotational Raman Coherence Spectroscopy with Femtosecond Pulses, pp. 1237ff. [11] Albert, S. et al. In Handbook of High-resolution Spectroscopy; Quack, M., Merkt, F., Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2011; Vol. 2; Chapter High-resolution Fourier Transform Infrared Spectroscopy, pp. 1153ff. [12] Weber, A. In Handbook of High-resolution Spectroscopy; Quack, M., Merkt, F., Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2011; Vol. 2; Chapter New Techniques in Microwave Spectroscopy, pp. 801ff.
We gratefully acknowledge funding support from the following institutions:
.
This work is supported by the National Research Foundation of Korea (NRF-2018R1D1A1A02042720) and Samsung Science and Technology Foundation (SSTF-BA2001-08). Past funding through Ulsan National Institute of Science and Technology (UNIST), Sonderforschungsbereich 450 of the Deutsche Forschungsgemeinschaft and the Max Born Institute in Berlin is grately acknowledged.